Proteomics Technologies
Proteomik
Customers and Partners benefit from Proteome Factory’s high quality contract research and fee-for-service in proteomics, protein quantification, identification, characterization and validation because of Proteome Factory's
- longtime know-how in proteomics, mass spectrometry, protein analysis and bioinformatics
- SOPs (standard operating procedures), quality management (ISO 9001 in progress)
- most modern and effective technology for extreme high resolution protein separation by 2DE, protein quantification and protein identification
- permanent innovations and advancements of technology platform
- highly automated protein identification
- highly reproducible proteome analysis
Comprehensive PFA-Proteomics Platform
Proteome Factory uses routinely several gel-based and gel-free proteomics approaches of its PFA-Proteomics Platform in contract research projects and internal R&D.
Depending on the scientific requirements the choice of the appropriate proteomics and protein analytical approach is discussed with clients and partners to ensure best possible proteomics studies. Since Proteome Factory’s foundation many proteome analysis studies and projects were performed for industrial and academic customers and internal R&D projects which were partially funded by grants of the ministry of science and education. Beside extreme high resolution 2D electrophoresis gel-free proteomics approaches with and without differential proteome protein labelling were used.
Proteomics
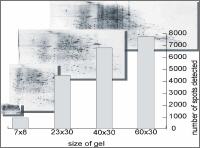
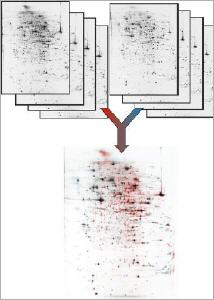
Extreme high resolution 2DE proteomics Depending on 2DE gel size up to theoretically 10000 protein spots can be separated within a 2DE gel.
Proteome Factory’s extreme high resolution 2DE gel electrophoresis platform increases the probability to find and identify high-valued biomarker and target proteins significantly. The 2DE platform is based on the invention of Professor Klose (Klose and Kobalz, Electrophoresis, 1995, 16, 1034-59) which is still the most powerful proteomics separation technique today. Depending on gel size several thousand up to 10,000 protein spots can be separated in one experiment. In combination with optimized sample prefractionation (e.g. depletion of abundant plasma proteins) and sample preparation of organs, tissue, cells in culture or body fluids Proteome Factory’s 2DE technique is superior compared to commercial available IPG gel based 2DE methods.
Depletion of high abundant proteins

Plasma 2DE proteomics with and without depletion of high abundance plasma proteins by affinity removal
Proteome analysis of plasma/serum and CSF is an important method to discover biomarkers and protein targets by comparison of disease- or therapy-associated samples with control samples which are the basis for development of new diagnostic tools and therapies.
Plasma, serum and CSF are the most difficult samples for differential proteomics. The dynamic concentration range of proteins is about 6 to 8 orders of magnitude. The portion of albumin is about 60-65% of the total protein amount followed by the fraction of immunglobulins (ca. 15%). The 20 most abundant proteins amount to ca. 99% of the total protein. Therefore it is highly recommendable to remove high abundant proteins from plasma or CSF by affinity depletion before proteome analysis to enhance the detection of low abundant proteins and penetrate deeper into the proteome.
Proteome Factory applies different affinity chromatographic methods for depletion of high abundant proteins which can increase the number of detectable protein spots on extreme high resolution 2DE gels by about 50% and more percent.
Automation in Proteomics
Proteome Factory uses automation in many parts of its PFA-Proteomics Platform to enhance reproducibility and throughput and to prevent contaminations for example by keratin.
The high throughput spot picking robot spotXpress cuts up to 800 protein spots per hour for protein identification or ultra sensitive highly reproducible quantification of MeCAT labelled proteins by ICP-MS.
SpotDigest 96 is a sample preparation robot for preparation of protein spots for enzymatic digestion and protein identification by mass spectrometry.
Protein identification by nanoLC-ESI-MSMS is performed fully automatic in 24/7 operation with periodical quality and sensitivity controls to ensure highest sensitivity and quality. Proteome Factory’s bioinformatics platform and PWB Protein Identification Cluster Solution enables full automatic protein identification by database searching and de novo sequencing of vast numbers of samples with peptide fragmentation fingerprint (PFF) MSMS data. The results are checked and validated by experienced scientists using different bioinformatical analysis methods.